# Rcwl/RcwlPipelines case study: scRNA-seq data preprocessing
# Introduction
Here we demonstrate a case study for scRNA-seq data preprocessing
using RcwlPipeline tools and pipeline.
10x Genomics has its own preprocessing pipeline Cell Ranger to
process the scRNA-seq outputs it produces to perform the
demultiplexing and quantification. However, it requires much
configuration to run and is significantly slower than other mappers.
In this case study,
STARsolo (opens new window)
is used for alignment and quantification, which produces a count
matrix from FASTQ.
DropletUtils (opens new window) is
used for filtering raw gene-barcode matrix and removing empty
droplets, which produces a high-quality count matrix with feature/cell
annotation files saved in an R object of SingleCellExperiment.
Before these, a one-time indexing step using
STARindex (opens new window)
is also included in this case study.
# scRNA-seq data source
The scRNA-seq data source is the 1k PBMCs from 10x genomics (These source files are provided in the Zenodo (opens new window) data repository).
The dataset used in this tutorial are sub-sampled from the source files to contain only 15 cells instead of 1000. The data curation is for demo purpose only so that the execution of the Rcwl scRNA-seq preprocessing tools or pipeline in R can be completed within 1~2 minutes.
The "*.fastq" data was curated to only include reads on chromosome 21.
“subset15_demo_barcode.txt” contains known cell barcodes for mapping and only 15 barcodes are included.
"Homo_sapiens.GRCh37.75.21.gtf" contains the hg19 GTF file to annotate reads, which was curated on chromosome 21 only.
Data can be loaded from the dedicated GitHub repository (opens new window) or the Zenodo (opens new window) data repository.
library(git2r)
clone("https://github.com/rworkflow/testdata", "rcwl_data_supp")
path <- "rcwl_data_supp" ## source data
dir(path)
Here we also create an output directory to save result files from running the tool/pipeline.
outpath <- "outdir"
dir.create(outpath, showWarnings = FALSE)
# Load the packages and sync all tools
# Install packages
If this is the first time you use Rcwl or RcwlPipelines, you need
to install the packages:
The package can be installed from Bioconductor (>= 3.9):
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("Rcwl", "RcwlPipelines"))
Or use the most updated version from GitHub:
BiocManager::install(c("rworkflow/Rcwl", "rworkflow/RcwlPipelines"))
Load the packages:
library(Rcwl)
library(RcwlPipelines)
# Load scRNA-seq pre-processing tools
Three core functions: cwlUpdate, cwlSearch and cwlLoadfrom
RcwlPipelines will be needed for updating, searching, and loading
the needed tools or pipelines in R.
The cwlUpdate function syncs the current Rcwl recipes and returns
a cwlHub object which contains the most updated Rcwl recipes. The
mcols() function returns all related information about each
available tool or pipeline. The recipes will be locally cached, so
users don't need to call cwlUpdate every time unless they want to
use a tool/pipeline that is newly added to RcwlPipelines.
In this example, we are using the Bioc 3.13 which is the current devel version.
atls <- cwlUpdate(branch = "dev", force = TRUE) ## sync the tools/pipelines.
atls
table(mcols(atls)$Type)
cwlSearch is used to search for specific tools/pipelines of
interest. Multiple keywords can be used for an internal search of
"rname", "rpath", "fpath", "Command" and "Containers" columns in the
mcols().
tls <- cwlSearch(c("STAR", "index"))
mcols(tls)
cwlLoad loads the Rcwl tool/pipeline into the R working
environment. Here we load all the 3 tools that will be needed for the
scRNA-seq data preprocessing. The recipes for developing these tools
can be found in the GitHub
repository (opens new window)
including
tl_STARindex (opens new window),
tl_STARsolo (opens new window),
and
tl_DropletUtils (opens new window).
STARindex <- cwlLoad(title(tls)[2]) ## "tl_STARindex"
## equivalent:
## STARindex <- cwlLoad(mcols(tls)$fpath[2])
STARsolo <- cwlLoad("tl_STARsolo")
DropletUtils <- cwlLoad("tl_DropletUtils")
# scRNA-seq preprocessing
# Indexing
Before read alignment and quality control, a one-time genome indexing
needs to be done. The command line using STAR will look like this:
$ STAR --runMode genomeGenerate --runThreadN 4 --genomeDir STARindex
--genomeFastaFiles chr21.fa --sjdbGTFfile Homo_sapiens.GRCh37.75.21.gtf
We can equivalently index the genome using the Rcwl tool of
STARindex within R, which was internally passed as cwl scripts, by
only assigning values to the input parameters, and execute the cwl
script using one of the execution functions, e.g., runCWL in the
local computer. Then the output files are ready to pass as input to
the next tool for single cell read alignment.
Assign values to the input parameters:
STARindex$genomeFastaFiles <- file.path(path, "chr21.fa")
STARindex$sjdbGTFfile <- file.path(path, "Homo_sapiens.GRCh37.75.21.gtf")
runCWL(cwl = STARindex, outdir = file.path(outpath, "STARindex_output"), docker = TRUE)
dir(file.path(outpath, "STARindex_output"), recursive = TRUE) ## output files
Note that the docker argument in runCWL function takes 4 values:
- TRUE (default, recommended), which automatically pulls docker images for the required command line tools.
- FALSE, if users have already pre-installed all required command line tools.
- "singularity" if the running environment doesn't support docker but singularity.
- "udocker" for the docker-like runtime without any administrator privileges.
# Alignment
Assign values to input parameters:
cdna.fastq <- file.path(path, list.files(path, pattern = "_R2_"))
cb.fastq <- file.path(path, list.files(path, pattern = "_R1_"))
cblist <- file.path(path, "subset15_demo_barcode.txt")
genomeDir <- file.path(outpath, "STARindex_output/STARindex")
inputs(STARsolo)
STARsolo$readFilesIn_cdna <- cdna.fastq
STARsolo$readFilesIn_cb <- cb.fastq
STARsolo$whiteList <- cblist
STARsolo$genomeDir <- genomeDir
runCWL(STARsolo, outdir = file.path(outpath, "STARsolo_output"))
dir(file.path(outpath, "STARsolo_output"), recursive = TRUE)
The output files generated in the "STARsolo_output" folder can now be passed into the next tool for QC.
# Count filtering
To get a high-quality count matrix we apply the DropletUtils Bioconductor package, which will produce a filtered dataset that is
more representative of the Cell Ranger pipeline.
Since CWL itself doesn't support the integration of R packages or
R function, this is a unique feature for Rcwl, where we can easily
connect the upstream data preprocessing steps (previously based on
command line tools) and the downstream data analysis steps (heavily
done in R/Bioconductor).
The idea here is to put anything you need into a user-defined R
function, with specified arguments for input and output files, then
it's ready to be wrapped as an Rcwl tools for execution.
For example, in wrapping the Bioconductor package
DropletUtils (opens new window)
functionalities, we wrote this Rcwl tool called
tl_DropletUtils (opens new window)
with 3 major steps: 1) use the read10xCounts function to read the
raw aligned files and convert into a SingleCellExperiment object. 2)
calculate the barcode ranks and plotting. 3) calculate the empty
droplets and plotting. We have also defined the tool output to collect
the SingleCellExperiment Rdata file and diagnostic figures pdf file.
library(DropletUtils)
inputs(DropletUtils)
DropletUtils$dirname <- file.path(outpath, "STARsolo_output/Solo.out")
DropletUtils$lower <- 100
DropletUtils$df <- 5
runCWL(DropletUtils, outdir = file.path(outpath, "dropletUtils_output"), showLog = FALSE)
dir(file.path(outpath, "dropletUtils_output"))
Now that we get 2 output files:
- The pdf file with 2 diagnostic figures: Barcode ranks, and empty droplets. For details regarding interpretation of each diagnostic figure, please refer to the DropletUtils vignette (opens new window).
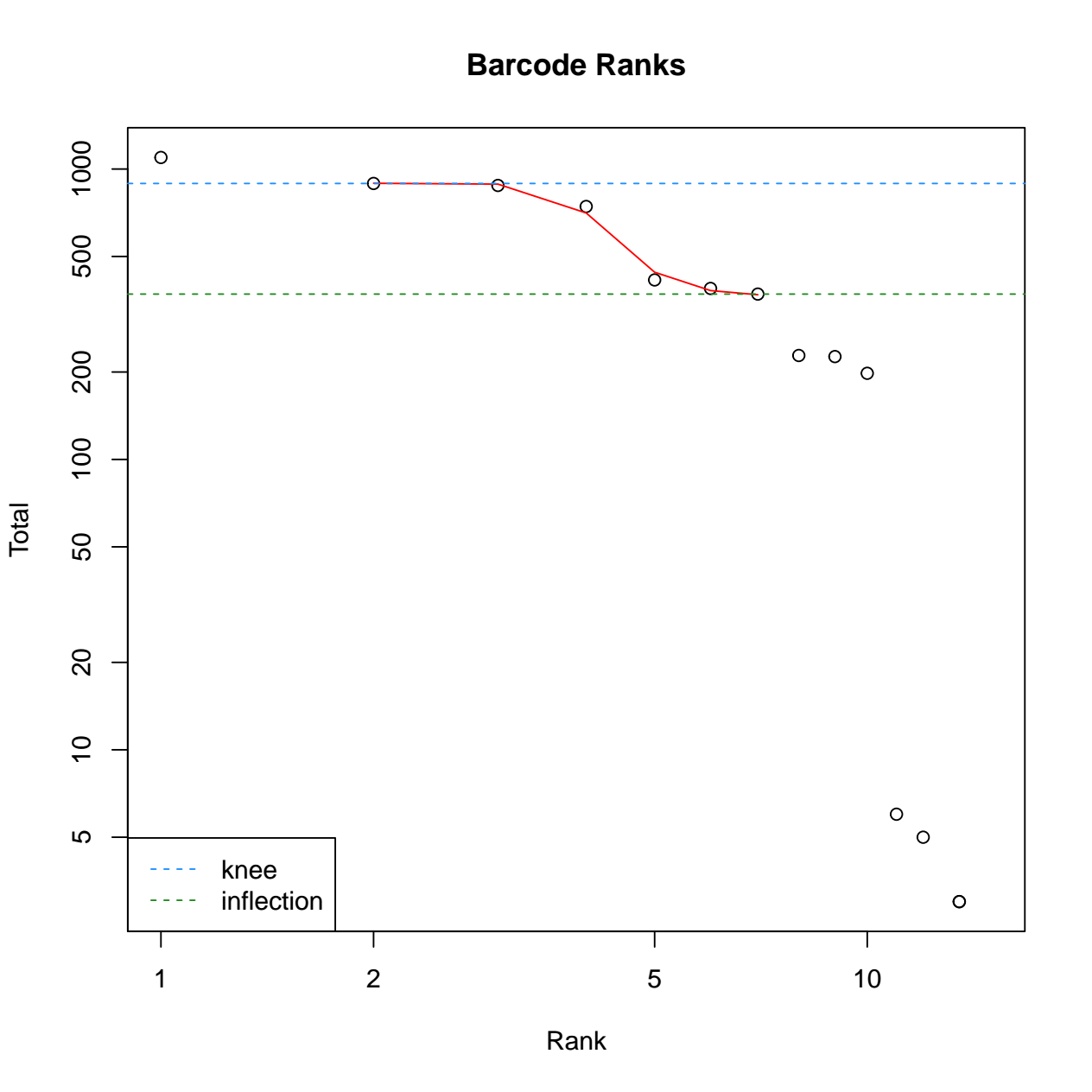
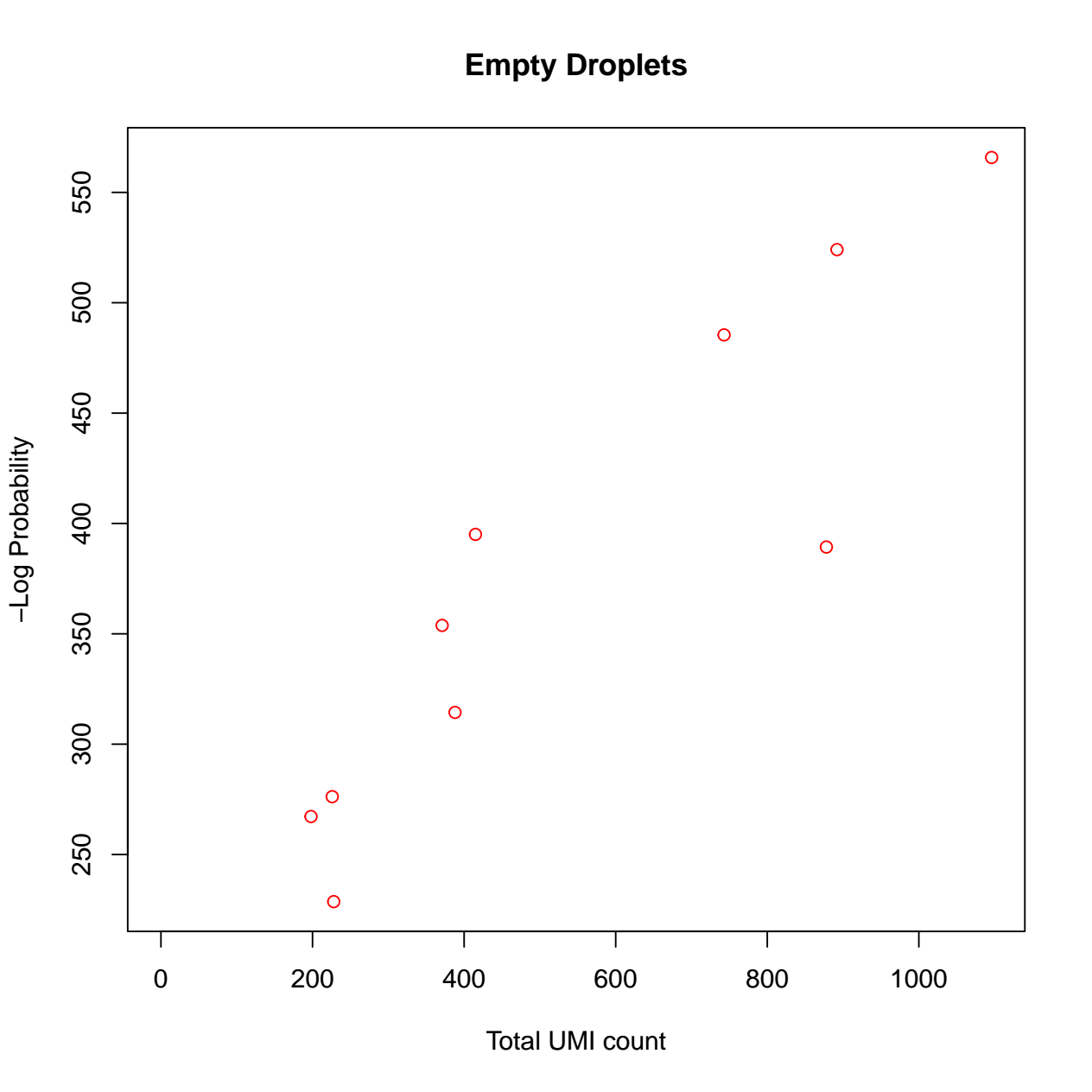
Biobase::openPDF(file.path(outpath, "dropletUtils_output/diagnostics.pdf"))
- The
SingleCellExperimentobject which has filtered out unqualified cells and is analysis ready.
sce <- readRDS(file.path(outpath, "dropletUtils_output/sce_filtered.rds"))
sce
# scRNA-seq preprocessing pipeline
Alternatively and more easily, we can use the pipeline called
pl_STARsoloDropletUtils (recipe
here (opens new window))
for the scRNA-seq data preprocessing. This pipeline integrated the
STARsolo and DropletUtils for a streamlined preprocessing analysis
within R. It is included in RcwlPipelines and is ready to be
customized for your own research. Pipelines can be visualized by the
plotCWL function.
STARsoloDropletUtils <- cwlLoad("pl_STARsoloDropletUtils")
plotCWL(STARsoloDropletUtils)
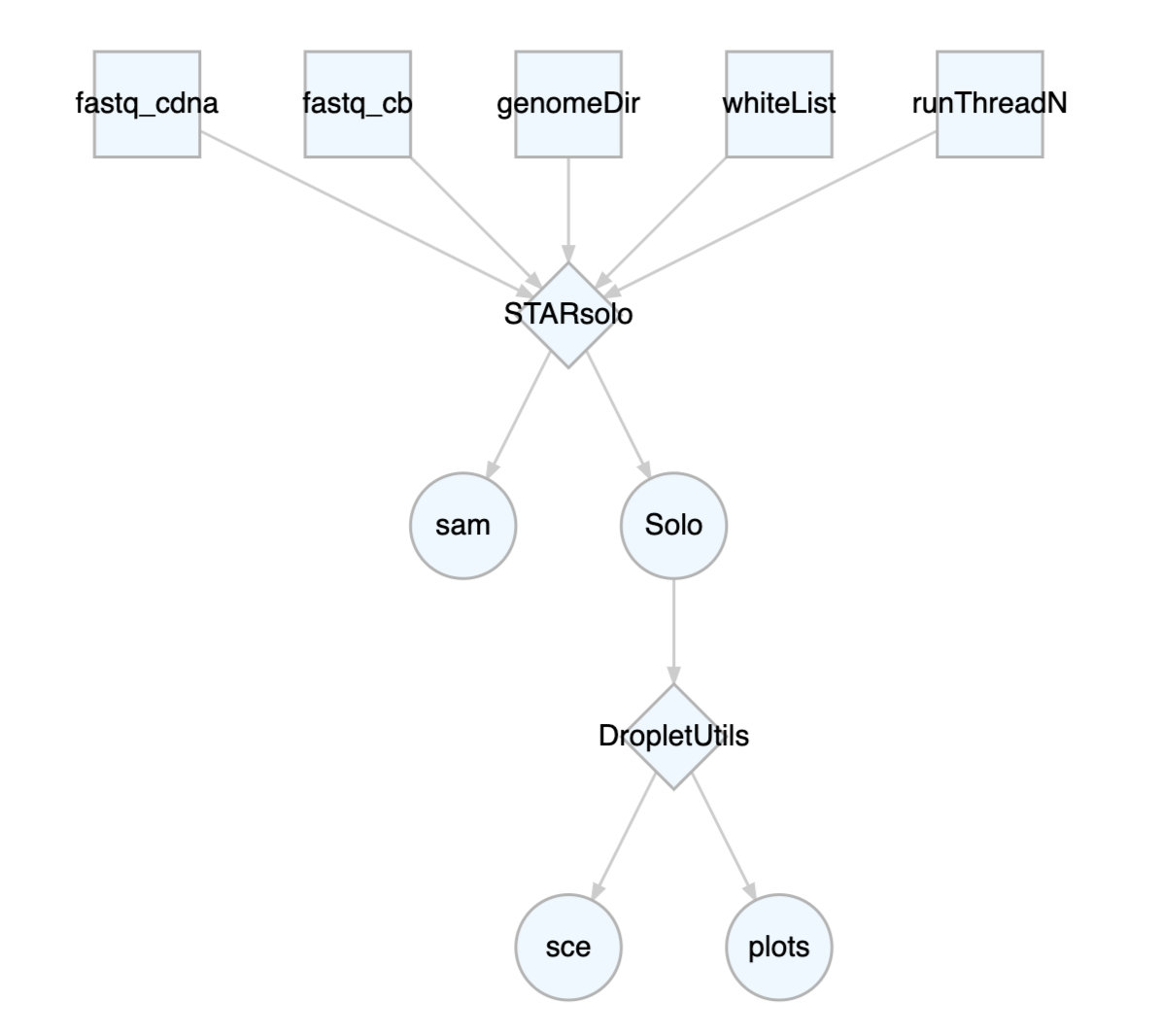
For Rcwl pipelines, we only need to assign input values for the
whole pipeline, not individual tools involved. The input and output
between each step are pre-defined in the pipeline to ensure a smooth
passing.
inputs(STARsoloDropletUtils)
STARsoloDropletUtils$fastq_cdna <- cdna.fastq
STARsoloDropletUtils$fastq_cb <- cb.fastq
STARsoloDropletUtils$genomeDir <- file.path(outpath, "STARindex_output/STARindex")
STARsoloDropletUtils$whiteList <- cblist
STARsoloDropletUtils$runThreadN <- 1
runCWL(STARsoloDropletUtils, outdir = file.path(outpath, "scpipeline_output"))
The overall output of the pipeline was pre-defined to glob the important files from separate steps.
outputs(STARsoloDropletUtils)
dir(file.path(outpath, "scpipeline_output"), recursive = TRUE)
# Submit parallel jobs for STARsolo
Powered by BiocParallel,Rcwl supports parallel job running for
multiple samples using the runCWLBatch function. The following
example demonstrates how to do the parallel alignment for the 2
samples using the STARsolo tool.
The BPPARAM argument in runCWLBatch() defines the parallel
parameters. It can be defined by BiocParallel::BatchtoolsParam
function, where the cluster argument takes different values for
different cluster job manager, such as "multicore", "sge" and
"slurm". More details about available options can be checked by
?BiocParallel::BatchtoolsParam.
library(BiocParallel)
bpparam <- BatchtoolsParam(workers = 2, cluster = "sge",
template = batchtoolsTemplate("sge"))
In the following example, we are using "multicore" for the parallel running.
The inputList argument is required to be a list of input parameter
values for samples that are to be computed parallelly. NOTE that
the names of the list must be consistent with the ids of input
parameters. In this example, the names are readFilesIn_cdna and
readFilesIn_cb.
The paramList argument is required to be a list of input parameter
values that are to be shared for all parallelly running samples.
bpparam <- BatchtoolsParam(workers = 2, cluster = "multicore")
input_lst <- list(readFilesIn_cdna = list(
sample1 = cdna.fastq,
sample2 = cdna.fastq),
readFilesIn_cb = list(
sample1 = cb.fastq,
sample2 = cb.fastq)
)
param_lst <- list(whiteList = cblist,
genomeDir = genomeDir,
runThreadN = 2)
res <- runCWLBatch(cwl = STARsolo,
outdir = file.path(outpath, "STARsolo_batch_output"),
inputList = input_lst, paramList = param_lst,
BPPARAM = bpparam)
The results are saved in separate folders for each parallel sample.
dir(file.path(outpath, "STARsolo_batch_output"), recursive = TRUE) ## output files
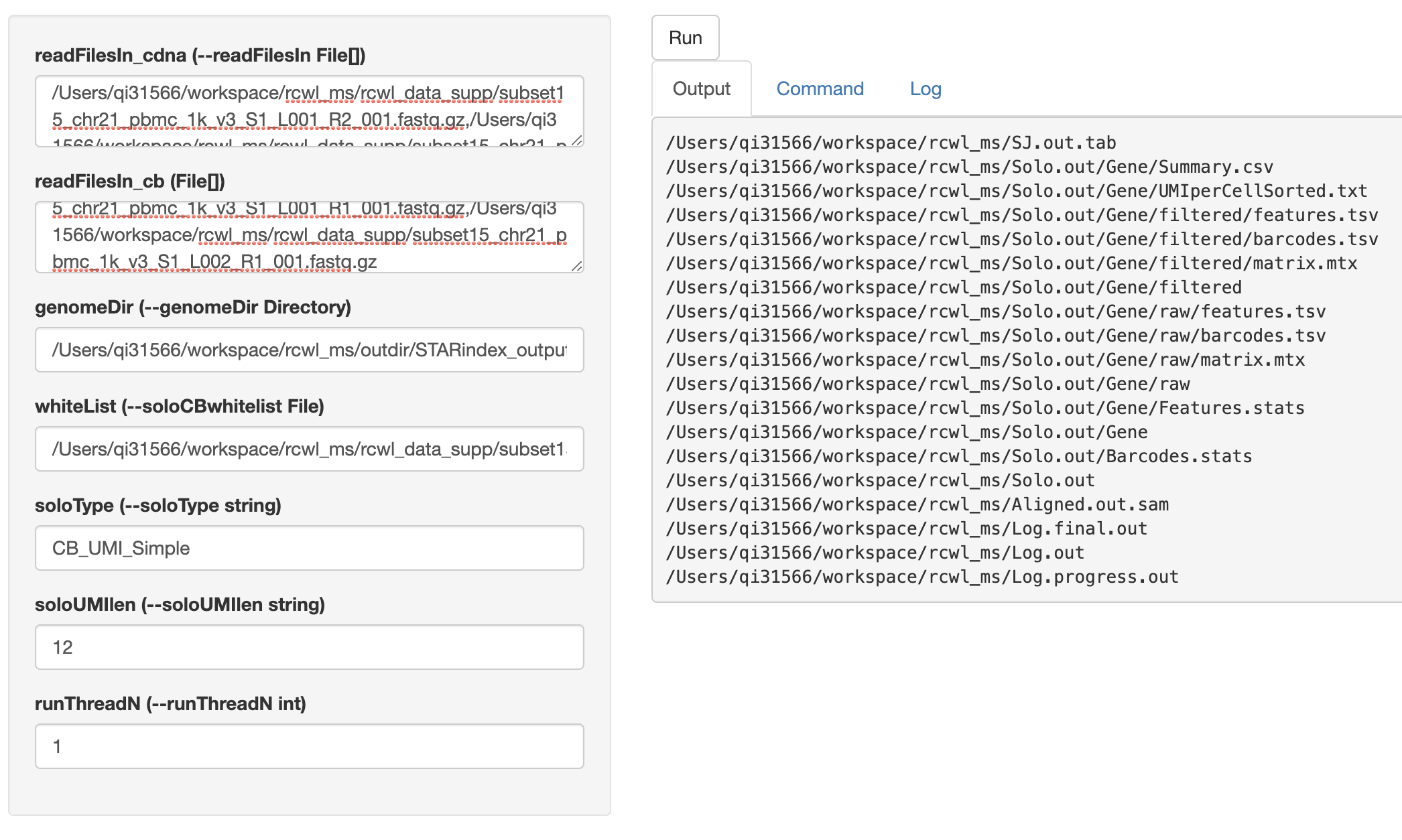
# Shiny interface
cwlShiny() opens a user-friendly shiny interface for running any
Rcwl tools or pipelines. By default, users need to put in the
absolute file path for each input parameter. NOTE, multiple file
paths need to be separated by colon. Click the run button, it will
start running and return the output file paths under Output
tag. Users can also check the Command and Log in the shiny
interface page.
cwlShiny(STARsolo)
# SessionInfo
sessionInfo()
# Links
- GitHub repository (opens new window) for the workshop materials and instructions.
- YouTube link (opens new window) for the Rcwl workshop on BioCAsia2020.