Rcwl: An R interface to the Common Workflow Language (CWL)
Qiang Hu, Qian Liu
2021-02-24
Rcwl.RmdHere we introduce the Bioconductor toolchain for usage and development of reproducible bioinformatics pipelines using packages of Rcwl and RcwlPipelines.
Rcwl provides a simple way to wrap command line tools and build CWL data analysis pipelines programmatically within R. It increases the ease of use, development, and maintenance of CWL pipelines. RcwlPipelines manages a collection of more than a hundred of pre-built and tested CWL tools and pipelines, which are highly modularized with easy customization to meet different bioinformatics data analysis needs.
In this vignette, we will introduce how to build and run CWL pipelines within R/Bioconductor using Rcwlpackage. More details about CWL can be found at https://www.commonwl.org.
Installation
- Download and install the package.
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("Rcwl")The development version with most up-to-date functionalities is also available from GitHub.
BiocManager::install("rworkflow/Rcwl")- Load the package into R session.
library(Rcwl)First Example
cwlProcess is the main constructor function to wrap a command line tool into an R tool as a cwlProcess object (S4 class). Let’s start with a simple example to wrap the echo command and execute echo hello world in R.
First, we need to define the input parameter for the base command echo, here it is a string without a prefix. An id argument is required here.
input1 <- InputParam(id = "sth")Second, we can construct a cwlProcess object by specifying the baseCommand for the command line tool, and InputParamList for the input parameters.
echo <- cwlProcess(baseCommand = "echo", inputs = InputParamList(input1))Now we have converted the command line tool echo into an R tool: an R object of class cwlProcess with the name of echo. We can take a look at the this R object and use some utility functions to extract specific information.
echo## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: echo
## inputs:
## sth (string):
## outputs:
## output:
## type: stdout
class(echo)## [1] "cwlProcess"
## attr(,"package")
## [1] "Rcwl"
cwlClass(echo)## [1] "CommandLineTool"
cwlVersion(echo)## [1] "v1.0"
baseCommand(echo)## [1] "echo"
inputs(echo)## inputs:
## sth (string):
outputs(echo)## outputs:
## output:
## type: stdoutThe inputs(echo) will show the value once it is assigned in next step. Since we didn’t define the outputs for this tool, it will stream standard output to a temporary file by default.
The third step is to assign values (here is “Hello World!”) for the input parameters.
echo$sth <- "Hello World!"
inputs(echo)## inputs:
## sth (string): Hello World!Now this R version of command line tool echo is ready to be executed.
The function runCWL runs the tools in R and returns a list of: 1) actual command line that was executed, 2) filepath to the output, and 3) running logs. The output directory by default takes the working directory, but can be specified in outdir argument.
## [1;30mINFO[0m Final process status is success
r1## List of length 3
## names(3): command output logs
r1$command## [1] "\033[1;30mINFO\033[0m [job echo.cwl] /private/tmp/docker_tmp4_wmpqno$ echo \\"
## [2] " 'Hello World!' > /private/tmp/docker_tmp4_wmpqno/30d6bd9a81a586a656cc7ca9ec98d1a692f05e40"
readLines(r1$output)## [1] "Hello World!"
r1$logs## [1] "\033[1;30mINFO\033[0m /Users/qi31566/Library/Python/3.7/bin/cwltool 3.0.20200324120055"
## [2] "\033[1;30mINFO\033[0m Resolved '/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T//Rtmpt8cgVU/file49d288ebeac/echo.cwl' to 'file:///var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/file49d288ebeac/echo.cwl'"
## [3] "\033[1;30mINFO\033[0m [job echo.cwl] /private/tmp/docker_tmp4_wmpqno$ echo \\"
## [4] " 'Hello World!' > /private/tmp/docker_tmp4_wmpqno/30d6bd9a81a586a656cc7ca9ec98d1a692f05e40"
## [5] "\033[1;30mINFO\033[0m [job echo.cwl] completed success"
## [6] "{"
## [7] " \"output\": {"
## [8] " \"location\": \"file:///var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/30d6bd9a81a586a656cc7ca9ec98d1a692f05e40\","
## [9] " \"basename\": \"30d6bd9a81a586a656cc7ca9ec98d1a692f05e40\","
## [10] " \"class\": \"File\","
## [11] " \"checksum\": \"sha1$a0b65939670bc2c010f4d5d6a0b3e4e4590fb92b\","
## [12] " \"size\": 13,"
## [13] " \"path\": \"/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/30d6bd9a81a586a656cc7ca9ec98d1a692f05e40\""
## [14] " }"
## [15] "}"
## [16] "\033[1;30mINFO\033[0m Final process status is success"Users can also have the log printed out by specifying showLog = TRUE.
## }A utility function writeCWL converts the cwlProcess object into 2 files: a .cwl file for the command and .yml file for the inputs, which are the internal cwl files to be executed when runCWL is invoked. The internal execution requires a cwl-runner (e.g., cwltool), which will be installed automatically with runCWL.
writeCWL(echo)## cwlout
## "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T//Rtmpt8cgVU/file49d263e42147/echo.cwl"
## ymlout
## "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T//Rtmpt8cgVU/file49d263e42147/echo.yml"Wrap command line tools
The package provides functions to define a CWL syntax for Command Line Tools in an intuitive way. The functions were developed based on the CWL Command Line Tool Description (v1.0). More details can be found in the official document: https://www.commonwl.org/v1.0/CommandLineTool.html.
Input Parameters
Essential Input parameters
For the input parameters, three options need to be defined usually, id, type, and prefix. The type can be string, int, long, float, double, and so on. More detail can be found at: https://www.commonwl.org/v1.0/CommandLineTool.html#CWLType.
Here is an example from CWL user guide. Here we defined an echo with different type of input parameters by InputParam. The stdout option can be used to capture the standard output stream to a file.
e1 <- InputParam(id = "flag", type = "boolean", prefix = "-f")
e2 <- InputParam(id = "string", type = "string", prefix = "-s")
e3 <- InputParam(id = "int", type = "int", prefix = "-i")
e4 <- InputParam(id = "file", type = "File", prefix = "--file=", separate = FALSE)
echoA <- cwlProcess(baseCommand = "echo",
inputs = InputParamList(e1, e2, e3, e4),
stdout = "output.txt")Then we give it a try by setting values for the inputs.
echoA$flag <- TRUE
echoA$string <- "Hello"
echoA$int <- 1
tmpfile <- tempfile()
write("World", tmpfile)
echoA$file <- tmpfile
r2 <- runCWL(echoA, outdir = tempdir())## [1;30mINFO[0m Final process status is success
r2$command## [1] "\033[1;30mINFO\033[0m [job echoA.cwl] /private/tmp/docker_tmp2xt4jr76$ echo \\"
## [2] " --file=/private/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/tmp0lp79zis/stg36f80227-107a-42f3-9a40-a5eef5e4bc6c/file49d227f75930 \\"
## [3] " -f \\"
## [4] " -i \\"
## [5] " 1 \\"
## [6] " -s \\"
## [7] " Hello > /private/tmp/docker_tmp2xt4jr76/output.txt"The command shows the parameters work as we defined. The parameter order is in alphabetical by default, but the option of “position” can be used to fix the orders.
Array Inputs
A similar example to CWL user guide. We can define three different type of array as inputs.
a1 <- InputParam(id = "A", type = "string[]", prefix = "-A")
a2 <- InputParam(id = "B",
type = InputArrayParam(items = "string",
prefix="-B=", separate = FALSE))
a3 <- InputParam(id = "C", type = "string[]", prefix = "-C=",
itemSeparator = ",", separate = FALSE)
echoB <- cwlProcess(baseCommand = "echo",
inputs = InputParamList(a1, a2, a3))Then set values for the three inputs.
echoB$A <- letters[1:3]
echoB$B <- letters[4:6]
echoB$C <- letters[7:9]
echoB## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: echo
## inputs:
## A (string[]): -A a b c
## B:
## type: array
## prefix: -B= d e f
## C (string[]): -C= g h i
## outputs:
## output:
## type: stdoutNow we can check whether the command behaves as we expected.
## [1;30mINFO[0m Final process status is success
r3$command## [1] "\033[1;30mINFO\033[0m [job echoB.cwl] /private/tmp/docker_tmpv4hszi02$ echo \\"
## [2] " -A \\"
## [3] " a \\"
## [4] " b \\"
## [5] " c \\"
## [6] " -B=d \\"
## [7] " -B=e \\"
## [8] " -B=f \\"
## [9] " -C=g,h,i > /private/tmp/docker_tmpv4hszi02/cf89575dc97103db92f466a558e5d0b8b42ec350"Output Parameters
Capturing Output
The outputs, similar to the inputs, is a list of output parameters. Three options id, type and glob can be defined. The glob option is used to define a pattern to find files relative to the output directory.
Here is an example to unzip a compressed gz file. First, we generate a compressed R script file.
zzfil <- file.path(tempdir(), "sample.R.gz")
zz <- gzfile(zzfil, "w")
cat("sample(1:10, 5)", file = zz, sep = "\n")
close(zz)We define a cwlProcess object to use “gzip” to uncompress a input file.
ofile <- "sample.R"
z1 <- InputParam(id = "uncomp", type = "boolean", prefix = "-d")
z2 <- InputParam(id = "out", type = "boolean", prefix = "-c")
z3 <- InputParam(id = "zfile", type = "File")
o1 <- OutputParam(id = "rfile", type = "File", glob = ofile)
gz <- cwlProcess(baseCommand = "gzip",
inputs = InputParamList(z1, z2, z3),
outputs = OutputParamList(o1),
stdout = ofile)Now the gz object can be used to uncompress the previous generated compressed file.
## [1;30mINFO[0m Final process status is success
r4$output## [1] "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/sample.R"Or we can use arguments to set some default parameters.
z1 <- InputParam(id = "zfile", type = "File")
o1 <- OutputParam(id = "rfile", type = "File", glob = ofile)
Gz <- cwlProcess(baseCommand = "gzip",
arguments = list("-d", "-c"),
inputs = InputParamList(z1),
outputs = OutputParamList(o1),
stdout = ofile)
Gz## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: gzip
## arguments: -d -c
## inputs:
## zfile (File):
## outputs:
## rfile:
## type: File
## outputBinding:
## glob: sample.R
## stdout: sample.R## [1;30mINFO[0m Final process status is successTo make it for general usage, we can define a pattern with javascript to glob the output, which require node from “nodejs” to be installed in your system PATH.
pfile <- "$(inputs.zfile.path.split('/').slice(-1)[0].split('.').slice(0,-1).join('.'))"Or we can use the CWL built in file property, nameroot, directly.
pfile <- "$(inputs.zfile.nameroot)"
o2 <- OutputParam(id = "rfile", type = "File", glob = pfile)
req1 <- requireJS()
GZ <- cwlProcess(baseCommand = "gzip",
arguments = list("-d", "-c"),
requirements = list(), ## assign list(req1) if node installed.
inputs = InputParamList(z1),
outputs = OutputParamList(o2),
stdout = pfile)
GZ$zfile <- zzfil
r4b <- runCWL(GZ, outdir = tempdir())## [1;30mINFO[0m Final process status is successArray Outputs
We can also capture multiple output files with glob pattern.
a <- InputParam(id = "a", type = InputArrayParam(items = "string"))
b <- OutputParam(id = "b", type = OutputArrayParam(items = "File"),
glob = "*.txt")
touch <- cwlProcess(baseCommand = "touch", inputs = InputParamList(a),
outputs = OutputParamList(b))
touch$a <- c("a.txt", "b.log", "c.txt")
r5 <- runCWL(touch, outdir = tempdir())## [1;30mINFO[0m Final process status is success
r5$output## [1] "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/a.txt"
## [2] "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/c.txt"The “touch” command generates three files, but the output only collects two files with “.txt” suffix as defined in the OutputParam using the “glob” option.
Running Tools in Docker
The CWL can work with docker to simplify your software management and communicate files between host and container. The docker container can be defined by the hints or requirements option.
d1 <- InputParam(id = "rfile", type = "File")
req1 <- requireDocker("r-base")
doc <- cwlProcess(baseCommand = "Rscript",
inputs = InputParamList(d1),
stdout = "output.txt",
hints = list(req1))
doc$rfile <- r4$output
r6 <- runCWL(doc)The tools defined with docker requirements can also be run locally by disabling the docker option. In case your Rscript depends some local libraries to run, an option from cwltools, “–preserve-entire-environment”, can be used to pass all environment variables.
## [1;30mINFO[0m Final process status is successRunning Tools in Cluster server
The CWL can also work in high performance clusters with batch-queuing system, such as SGE, PBS, SLURM and so on, using the Bioconductor package BiocParallel. Here is an example to submit jobs with “Multicore” and “SGE”.
library(BiocParallel)
sth.list <- as.list(LETTERS)
names(sth.list) <- LETTERS
## submit with multicore
result1 <- runCWLBatch(cwl = echo, outdir = tempdir(), inputList = list(sth = sth.list),
BPPARAM = MulticoreParam(26))
## submit with SGE
result2 <- runCWLBatch(cwl = echo, outdir = tempdir(), inputList = list(sth = sth.list),
BPPARAM = BatchtoolsParam(workers = 26, cluster = "sge",
resources = list(queue = "all.q")))Writing Pipeline
We can connect multiple tools together into a pipeline. Here is an example to uncompress an R script and execute it with Rscript.
Here we define a simple Rscript tool without using docker.
d1 <- InputParam(id = "rfile", type = "File")
Rs <- cwlProcess(baseCommand = "Rscript",
inputs = InputParamList(d1))
Rs## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: Rscript
## inputs:
## rfile (File):
## outputs:
## output:
## type: stdoutTest run:
## [1;30mINFO[0m Final process status is success
readLines(tres$output)## [1] "[1] 9 2 3 5 10"The pipeline includes two steps, decompressing with predefined cwlProcess of GZ and compiling with cwlProcess of Rs. The input file is a compressed file for the first “Uncomp” step.
i1 <- InputParam(id = "cwl_zfile", type = "File")
s1 <- cwlStep(id = "Uncomp", run = GZ,
In = list(zfile = "cwl_zfile"))
s2 <- cwlStep(id = "Compile", run = Rs,
In = list(rfile = "Uncomp/rfile"))In step 1 (‘s1’), the pipeline runs the cwlProcess of GZ, where the input zfile is defined in ‘i1’ with id of “cwl_zfile”. In step 2 (‘s2’), the pipeline runs the cwlProcess of Rs, where the input rfile is from the output of the step 1 (“Uncomp/rfile”) using the format of <step>/<output>.
The pipeline output will be defined as the output of the step 2 (“Compile/output”) using the format of <step>/<output> as shown below.
o1 <- OutputParam(id = "cwl_cout", type = "File",
outputSource = "Compile/output")The cwlWorkflow function is used to initiate the pipeline by specifying the inputs and outputs. Then we can simply use + to connect all steps to build the final pipeline.
cwl <- cwlWorkflow(inputs = InputParamList(i1),
outputs = OutputParamList(o1))
cwl <- cwl + s1 + s2
cwl## class: cwlWorkflow
## cwlClass: Workflow
## cwlVersion: v1.0
## inputs:
## cwl_zfile (File):
## outputs:
## cwl_cout:
## type: File
## outputSource: Compile/output
## steps:
## Uncomp:
## run: Uncomp.cwl
## in:
## zfile: cwl_zfile
## out:
## - rfile
## Compile:
## run: Compile.cwl
## in:
## rfile: Uncomp/rfile
## out:
## - outputLet’s run the pipeline.
## [1;30mINFO[0m Final process status is success
readLines(r7$output)## [1] "[1] 7 10 2 6 1"Tips: Sometimes, we need to adjust some arguments of certain tools in a pipeline besides of parameter inputs. The function arguments can help to modify arguments for a tool, tool in a pipeline, or even tool in a sub-workflow. For example,
## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: gzip
## arguments: -d -c -f
## inputs:
## zfile (File): /private/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/sample.R.gz
## outputs:
## rfile:
## type: File
## outputBinding:
## glob: $(inputs.zfile.nameroot)
## stdout: $(inputs.zfile.nameroot)Scattering pipeline
The scattering feature can specifies the associated workflow step or subworkflow to execute separately over a list of input elements. To use this feature, ScatterFeatureRequirement must be specified in the workflow requirements. Different scatter methods can be used in the associated step to decompose the input into a discrete set of jobs. More details can be found at: https://www.commonwl.org/v1.0/Workflow.html#WorkflowStep.
Here is an example to execute multiple R scripts. First, we need to set the input and output types to be array of “File”, and add the requirements. In the “Compile” step, the scattering input is required to be set with the scatter option.
i2 <- InputParam(id = "cwl_rfiles", type = "File[]")
o2 <- OutputParam(id = "cwl_couts", type = "File[]", outputSource = "Compile/output")
req1 <- requireScatter()
cwl2 <- cwlWorkflow(requirements = list(req1),
inputs = InputParamList(i2),
outputs = OutputParamList(o2))
s1 <- cwlStep(id = "Compile", run = Rs,
In = list(rfile = "cwl_rfiles"),
scatter = "rfile")
cwl2 <- cwl2 + s1
cwl2## class: cwlWorkflow
## cwlClass: Workflow
## cwlVersion: v1.0
## requirements:
## - class: ScatterFeatureRequirement
## inputs:
## cwl_rfiles (File[]):
## outputs:
## cwl_couts:
## type: File[]
## outputSource: Compile/output
## steps:
## Compile:
## run: Compile.cwl
## in:
## rfile: cwl_rfiles
## out:
## - output
## scatter: rfileMultiple R scripts can be assigned to the workflow inputs and executed.
## [1;30mINFO[0m Final process status is success
r8$output## [1] "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/d197503c167721206ef2fc1631cd2a8b6c9c16d7"
## [2] "/var/folders/7t/9l4kkf_j2sqbpn321y9g5558z96ck_/T/Rtmpt8cgVU/d197503c167721206ef2fc1631cd2a8b6c9c16d7_2"Pipeline plot
The function plotCWL can be used to visualize the relationship of inputs, outputs and the analysis for a tool or pipeline.
plotCWL(cwl)## Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if `.name_repair` is omitted as of tibble 2.0.0.
## Using compatibility `.name_repair`.Web Application
cwlProcess example
Here we build a tool with different types of input parameters.
e1 <- InputParam(id = "flag", type = "boolean",
prefix = "-f", doc = "boolean flag")
e2 <- InputParam(id = "string", type = "string", prefix = "-s")
e3 <- InputParam(id = "option", type = "string", prefix = "-o")
e4 <- InputParam(id = "int", type = "int", prefix = "-i", default = 123)
e5 <- InputParam(id = "file", type = "File",
prefix = "--file=", separate = FALSE)
e6 <- InputParam(id = "array", type = "string[]", prefix = "-A",
doc = "separated by comma")
mulEcho <- cwlProcess(baseCommand = "echo", id = "mulEcho",
label = "Test parameter types",
inputs = InputParamList(e1, e2, e3, e4, e5, e6),
stdout = "output.txt")
mulEcho## class: cwlProcess
## cwlClass: CommandLineTool
## cwlVersion: v1.0
## baseCommand: echo
## inputs:
## flag (boolean): -f
## string (string): -s
## option (string): -o
## int (int): -i 123
## file (File): --file=
## array (string[]): -A
## outputs:
## output:
## type: stdout
## stdout: output.txtcwlProcess to Shiny App
Some input parameters can be predefined in a list, which will be converted to select options in the webapp. An upload parameter can be used to defined whether to generate an upload interface for the file type option. If FALSE, the upload field will be text input (file path) instead of file input.
inputList <- list(option = c("option1", "option2"))
app <- cwlShiny(mulEcho, inputList, upload = TRUE)
runApp(app)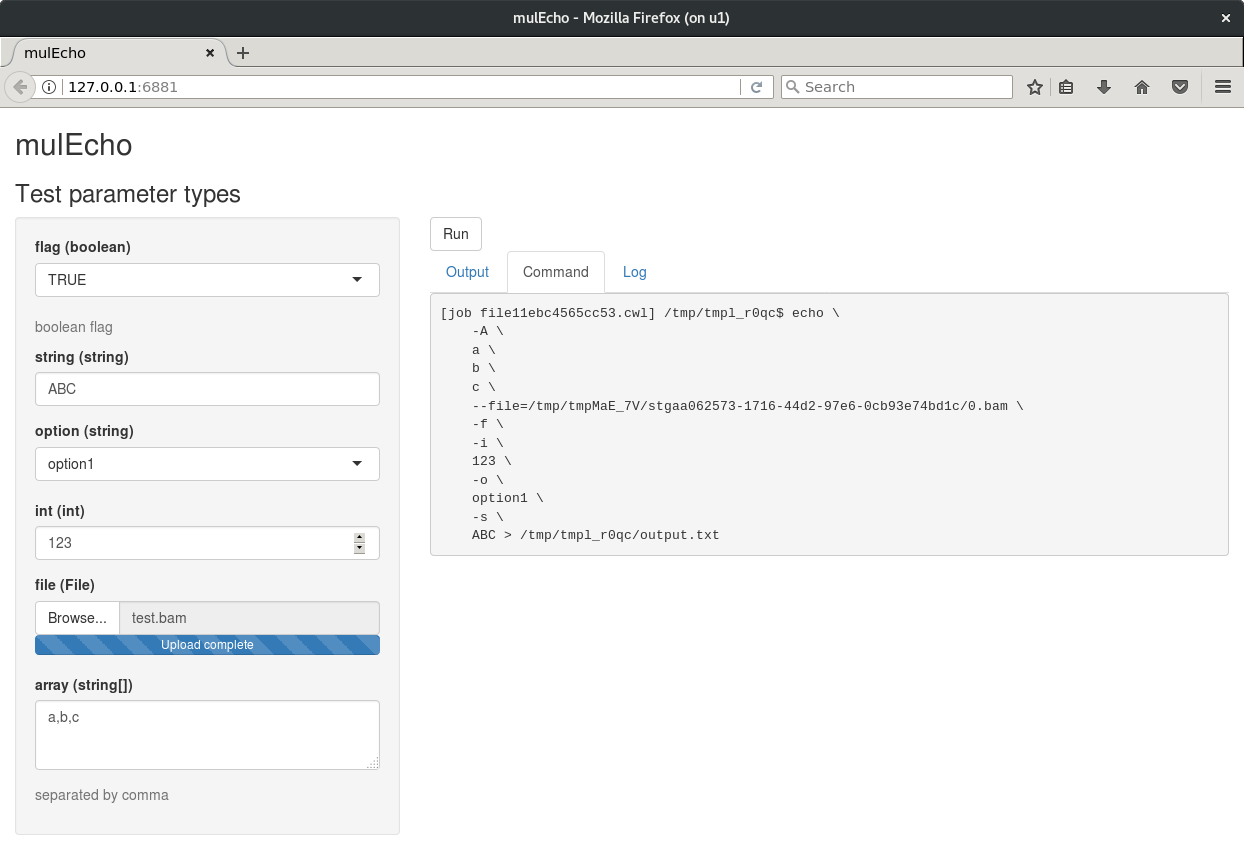
shinyApp
Working with R functions
We can wrap an R function to cwlProcess object by simply assigning the R function to baseCommand. This could be useful to summarize results from other tools in a pipeline. It can also be used to benchmark different parameters for a method written in R. Please note that this feature is only implemented by Rcwl, but not available in the common workflow language.
fun1 <- function(x)x*2
testFun <- function(a, b){
cat(fun1(a) + b^2, sep="\n")
}
assign("fun1", fun1, envir = .GlobalEnv)
assign("testFun", testFun, envir = .GlobalEnv)
p1 <- InputParam(id = "a", type = "int", prefix = "a=", separate = F)
p2 <- InputParam(id = "b", type = "int", prefix = "b=", separate = F)
o1 <- OutputParam(id = "o", type = "File", glob = "rout.txt")
TestFun <- cwlProcess(baseCommand = testFun,
inputs = InputParamList(p1, p2),
outputs = OutputParamList(o1),
stdout = "rout.txt")
TestFun$a <- 1
TestFun$b <- 2
r1 <- runCWL(TestFun, cwlArgs = "--preserve-entire-environment")## [1;30mINFO[0m Final process status is success
readLines(r1$output)## [1] "6"The runCWL function wrote the testFun function and its dependencies into an R script file automatically and call Rscript to run the script with parameters. Each parameter requires a prefix from corresponding argument in the R function with “=” and without a separator. Here we assigned the R function and its dependencies into the global environment because it will start a new environment when the vignette is compiled.
Resources
RcwlPipelines
The Rcwl package can be utilized to develop pipelines for best practices of reproducible research, especially for Bioinformatics study. Multiple Bioinformatics pipelines, such as RNA-seq alignment, quality control and quantification, DNA-seq alignment and variant calling, have been developed based on the tool in an R package RcwlPipelines, which contains the CWL recipes and the scripts to create the pipelines. Examples to analyze real data are also included.
The package is currently available in GitHub.
To install the package.
BiocManager::install("rworkflow/RcwlPipelines")The project website https://rcwl.org/ serves as a central hub for all related resources. It provides guidance for new users and tutorials for both users and developers. Specific resources are listed below.
Tutorial book
The tutorial book provides detailed instructions for developing Rcwl tools/pipelines, and also includes examples of some commonly-used tools and pipelines that covers a wide range of Bioinformatics data analysis needs.
The R recipes and cwl scripts
The R scripts to build the CWL tools and pipelines are now residing in a dedicated GitHub repository, which is intended to be a community effort to collect and contribute Bioinformatics tools and pipelines using Rcwl and CWL.
Tool collections in CWL format
Plenty of Bioinformatics tools and workflows can be found from GitHub in CWL format. They can be imported to cwlProcess object by readCWL function, or can be used directly.
SessionInfo
## R version 4.0.3 (2020-10-10)
## Platform: x86_64-apple-darwin13.4.0 (64-bit)
## Running under: macOS Catalina 10.15.7
##
## Matrix products: default
## BLAS/LAPACK: /Users/qi31566/miniconda3/envs/r-base/lib/libopenblasp-r0.3.12.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] Rcwl_1.7.12 S4Vectors_0.28.1 BiocGenerics_0.36.0
## [4] yaml_2.2.1 BiocStyle_2.18.1
##
## loaded via a namespace (and not attached):
## [1] tidyr_1.1.2 sass_0.3.1 jsonlite_1.7.2
## [4] R.utils_2.10.1 bslib_0.2.4 shiny_1.6.0
## [7] assertthat_0.2.1 BiocManager_1.30.10 base64url_1.4
## [10] progress_1.2.2 pillar_1.5.0 backports_1.2.1
## [13] lattice_0.20-41 glue_1.4.2 reticulate_1.18
## [16] digest_0.6.27 RColorBrewer_1.1-2 promises_1.2.0.1
## [19] checkmate_2.0.0 htmltools_0.5.1.1 httpuv_1.5.5
## [22] Matrix_1.3-2 R.oo_1.24.0 pkgconfig_2.0.3
## [25] bookdown_0.21 DiagrammeR_1.0.6.1 purrr_0.3.4
## [28] xtable_1.8-4 brew_1.0-6 later_1.1.0.1
## [31] BiocParallel_1.24.1 tibble_3.0.6 generics_0.1.0
## [34] ellipsis_0.3.1 cachem_1.0.4 withr_2.4.1
## [37] magrittr_2.0.1 crayon_1.4.1 mime_0.10
## [40] memoise_2.0.0 evaluate_0.14 R.methodsS3_1.8.1
## [43] fs_1.5.0 fansi_0.4.2 textshaping_0.3.1
## [46] tools_4.0.3 data.table_1.14.0 prettyunits_1.1.1
## [49] hms_1.0.0 lifecycle_1.0.0 basilisk.utils_1.2.2
## [52] stringr_1.4.0 compiler_4.0.3 pkgdown_1.6.1
## [55] jquerylib_0.1.3 systemfonts_1.0.1 rlang_0.4.10
## [58] grid_4.0.3 rstudioapi_0.13 rappdirs_0.3.3
## [61] htmlwidgets_1.5.3 visNetwork_2.0.9 igraph_1.2.6
## [64] rmarkdown_2.7 basilisk_1.2.1 codetools_0.2-18
## [67] DBI_1.1.1 R6_2.5.0 knitr_1.31
## [70] dplyr_1.0.4 fastmap_1.1.0 utf8_1.1.4
## [73] filelock_1.0.2 rprojroot_2.0.2 ragg_1.1.0
## [76] desc_1.2.0 stringi_1.5.3 Rcpp_1.0.6
## [79] vctrs_0.3.6 batchtools_0.9.15 tidyselect_1.1.0
## [82] xfun_0.21